Gene Therapy’s Role in Sanfilippo Syndrome: Latest Studies and Breakthroughs
Sanfilippo syndrome, also known as mucopolysaccharidosis type III (MPS III), represents one of the most devastating lysosomal storage disorders affecting children worldwide. This rare genetic condition results from deficiency in enzymes responsible for breaking down heparan sulfate, a complex sugar molecule found throughout the body. Without proper enzymatic function, these molecules accumulate in cells, particularly in the brain and nervous system, leading to progressive neurological decline, cognitive deterioration, and severe behavioral changes. Families facing this diagnosis have historically encountered limited treatment options, making the emergence of gene therapy approaches a transformative development in pediatric medicine.
The landscape of Sanfilippo syndrome treatment has undergone remarkable transformation in recent years. Where once management consisted primarily of supportive care and symptom management, innovative therapeutic approaches now offer genuine hope for disease modification. Gene therapy represents the most promising frontier, with multiple clinical trials demonstrating the ability to halt disease progression and, in some cases, reverse existing neurological damage. This comprehensive exploration examines the current state of gene therapy for Sanfilippo syndrome, reviewing the latest scientific evidence, clinical trial outcomes, and the mechanisms through which these revolutionary treatments work.
Understanding Sanfilippo Syndrome and Its Genetic Basis
Sanfilippo syndrome emerges from mutations in genes encoding specific lysosomal enzymes responsible for heparan sulfate degradation. The condition encompasses four distinct subtypes—A, B, C, and D—each resulting from deficiency in different enzymes within the heparan sulfate degradation pathway. Type A, the most common and typically most severe form, stems from mutations in the SGSH gene, which codes for heparan sulfate-specific heparitidase 1. Type B involves NAGLU gene mutations affecting N-acetyl-alpha-D-glucosaminidase. Type C results from HGSNAT gene mutations, while Type D involves GNS gene defects.
The pathophysiology of Sanfilippo syndrome involves progressive accumulation of undegraded heparan sulfate within lysosomes, particularly affecting neurons and glial cells. This accumulation triggers cellular dysfunction, neuroinflammation, mitochondrial impairment, and ultimately neuronal death. Clinical manifestations typically begin between ages two and six, with initial signs including developmental delay, behavioral problems, and sleep disturbances. As the disease progresses, children experience severe cognitive decline, loss of motor function, seizures, and eventually complete neurological deterioration. The progressive nature and lack of effective treatments historically made Sanfilippo syndrome uniformly fatal, with most patients dying in their teenage years.
Understanding this genetic foundation proved essential for developing targeted gene therapy approaches. Researchers recognized that delivering functional copies of deficient genes directly to affected cells, particularly neurons, could restore enzymatic activity and halt the disease process. This insight catalyzed the development of multiple gene therapy platforms, each employing different delivery mechanisms and targeting strategies to achieve therapeutic benefit.
Gene Therapy Mechanisms and Delivery Systems
Gene therapy for Sanfilippo syndrome operates through several distinct mechanisms, each designed to restore enzymatic function and prevent heparan sulfate accumulation. The primary approach involves in vivo gene delivery, where therapeutic genetic material is introduced directly into patients’ bodies, typically targeting the central nervous system where neurological damage occurs. Researchers have explored multiple delivery vehicles, with adeno-associated viruses (AAVs) emerging as the most clinically advanced platform for treating Sanfilippo syndrome.
AAV-based vectors offer several advantages for CNS gene delivery. These naturally occurring viruses have evolved mechanisms for crossing the blood-brain barrier and transducing neurons, making them ideal vehicles for delivering therapeutic genes to affected brain tissue. Researchers have engineered AAV variants with enhanced BBB penetration and neuronal tropism, improving therapeutic efficacy. The relatively small genome size of AAVs requires truncation of target genes, necessitating the development of self-complementary AAV vectors and split-intein approaches to overcome packaging constraints.
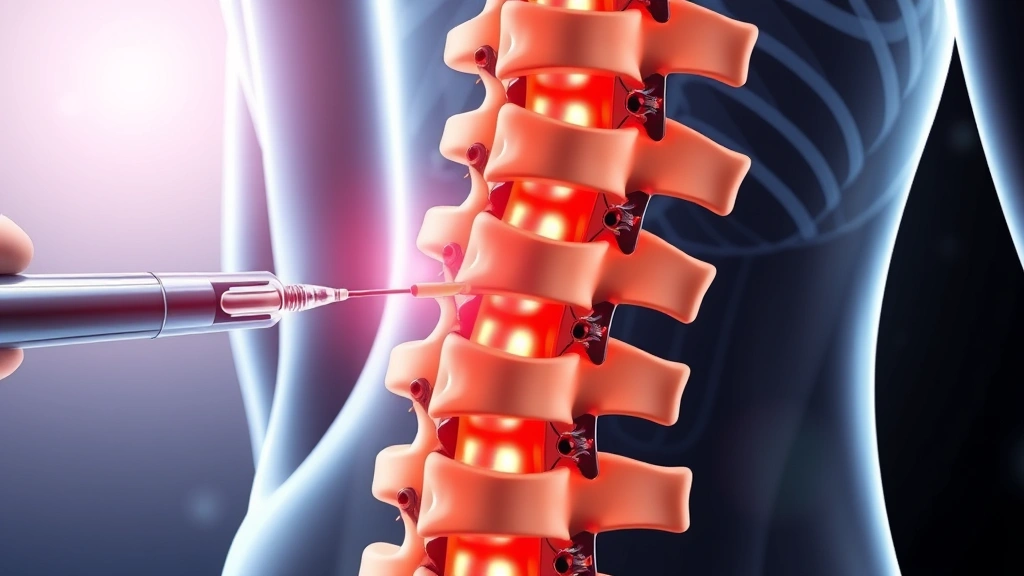
An alternative delivery mechanism involves intrathecal injection, where gene therapy vectors are administered directly into the cerebrospinal fluid surrounding the spinal cord and brain. This approach achieves high local concentrations of therapeutic vectors while minimizing systemic exposure. Several clinical trials have employed intrathecal delivery, demonstrating efficacy in halting disease progression and improving neurological outcomes. The direct CNS route proves particularly valuable for Sanfilippo syndrome, where the blood-brain barrier presents a significant obstacle to systemic gene delivery.
Speech and language therapists increasingly collaborate with gene therapy specialists to assess and monitor neurological function in treated patients. These professionals provide valuable outcome measures documenting improvements in communication abilities and cognitive function following treatment.
Clinical Trial Results and Patient Outcomes
Recent clinical trials investigating gene therapy for Sanfilippo syndrome have produced remarkably encouraging results, fundamentally changing the disease prognosis for treated patients. The most significant breakthrough emerged from trials of HT-100, an intrathecally delivered gene therapy for Sanfilippo syndrome Type A. Published data demonstrated that treated patients experienced stabilization of cognitive decline and, in some cases, measurable improvements in neurological function.
A pivotal Phase 1/2 trial enrolled young children with Sanfilippo Type A and assessed safety, tolerability, and efficacy over extended follow-up periods. Results revealed that treated patients maintained cognitive function at levels significantly better than untreated natural history controls. Standardized cognitive assessments showed that while untreated children experienced progressive decline, treated patients either maintained baseline function or demonstrated modest improvements. Furthermore, biomarker analysis revealed reduced levels of heparan sulfate in cerebrospinal fluid, indicating successful restoration of enzymatic activity and substrate clearance.
Additional clinical trials targeting different Sanfilippo subtypes have similarly demonstrated disease-modifying effects. Patients treated with gene therapy show improvements in behavioral symptoms, sleep disturbances, and motor function. Neuroimaging studies reveal preservation of brain structure and connectivity in treated patients compared to untreated controls. These objective measures complement subjective reports from families describing improved alertness, better social engagement, and enhanced quality of life in treated children.
Long-term follow-up data extending beyond five years post-treatment provides additional confidence in the durability and safety of gene therapy approaches. Treated patients continue demonstrating stable or improved neurological function, with no serious late adverse events attributable to the therapy. This extended follow-up proves particularly important for establishing gene therapy as a definitive disease-modifying treatment rather than a temporary intervention.
Physical therapy interventions complement gene therapy, with specialized rehabilitation professionals helping treated patients maximize functional recovery and maintain physical abilities. The combination of genetic correction with intensive rehabilitation often produces superior outcomes compared to either approach alone.
Different Subtypes and Targeted Approaches
The heterogeneity of Sanfilippo syndrome, with four distinct genetic subtypes, necessitates development of subtype-specific gene therapy approaches. While Type A has received the most clinical attention, recent progress has advanced treatments for Types B, C, and D. This comprehensive approach recognizes that each subtype requires delivery of a different therapeutic gene and may benefit from distinct delivery strategies optimized for each enzyme’s characteristics.
Sanfilippo Type B, caused by NAGLU gene mutations, represents the second most common variant. Gene therapy trials for Type B employ similar intrathecal delivery platforms but carrying the NAGLU gene rather than SGSH. Early clinical data from Type B trials demonstrates comparable disease modification to Type A approaches, with treated patients showing stabilization of cognitive decline and improvements in behavioral symptoms. The success of Type B trials validates the broader gene therapy platform and demonstrates its applicability across different genetic subtypes.
Type C Sanfilippo syndrome, resulting from HGSNAT mutations, presents unique challenges due to the gene’s larger size and the enzyme’s distinct subcellular localization. Researchers have developed innovative split-gene and dual-vector approaches to overcome packaging constraints while ensuring proper enzyme trafficking. Early preclinical and clinical data suggest these approaches successfully restore enzymatic function and prevent substrate accumulation.
Type D, the rarest subtype, has received less clinical attention but remains an important target for gene therapy development. Ongoing preclinical studies are establishing optimal delivery strategies and validating therapeutic efficacy in disease models. As gene therapy platforms mature, Type D patients will increasingly access treatments comparable in effectiveness to those available for more common subtypes.
This multi-subtype approach reflects broader recognition that rare genetic diseases require customized therapeutic strategies. Regulatory agencies and pharmaceutical companies increasingly support development of therapies targeting specific genetic subtypes, even when patient populations are small. This paradigm shift has accelerated progress in Sanfilippo syndrome treatment and offers hope to families affected by rarer disease variants.
Current Treatment Landscape and Approved Therapies
The regulatory landscape for Sanfilippo syndrome gene therapy has evolved dramatically, with multiple therapies advancing toward approval or achieving regulatory authorization. Zolgensma-like approaches, demonstrating success in spinal muscular atrophy, have informed gene therapy development for Sanfilippo syndrome. These precedents established regulatory pathways and clinical trial designs now being applied to mucopolysaccharidosis treatments.
Current approved and investigational therapies include intrathecal gene therapy products demonstrating disease-modifying effects in clinical trials. FDA regulatory reviews have accelerated for promising Sanfilippo syndrome treatments, recognizing the severe unmet medical need and compelling clinical evidence. Several therapies have achieved breakthrough therapy designation, expediting development timelines and enabling more frequent regulatory interactions.
Beyond gene therapy, complementary approaches including enzyme replacement therapy and substrate reduction therapy continue advancing. However, gene therapy’s ability to achieve durable disease modification through single or limited dosing makes it the most promising long-term treatment strategy. The combination of gene therapy with supportive care, rehabilitation, and emerging symptom management approaches creates a comprehensive treatment paradigm offering genuine hope for affected families.
Access to these emerging therapies remains challenging for many families, particularly those in regions with limited healthcare infrastructure or insurance coverage limitations. Advocacy organizations, patient support networks, and pharmaceutical companies increasingly collaborate to ensure equitable access and provide comprehensive support to families navigating gene therapy treatment decisions.
Occupational therapy professionals play essential roles in supporting gene therapy patients, helping them optimize daily functioning and adapt to neurological changes. These specialists provide valuable functional assessments documenting improvements following gene therapy initiation.
Challenges and Future Directions
Despite remarkable progress, significant challenges remain in optimizing gene therapy for Sanfilippo syndrome. The blood-brain barrier continues presenting obstacles to systemic gene delivery, necessitating invasive intrathecal administration. Researchers are actively developing novel delivery technologies including engineered AAV variants with enhanced BBB penetration, lipid nanoparticles, and other innovative approaches that might enable less invasive administration routes.
The optimal timing of gene therapy administration represents another critical consideration. Early treatment before extensive neurological damage occurs appears to yield superior outcomes, yet early diagnosis remains challenging. Expanded newborn screening programs could identify affected infants before symptom onset, enabling preventive gene therapy administration. This paradigm shift from treating symptomatic patients to preventing disease through early intervention represents an important future direction.
Long-term durability of gene therapy effects requires ongoing investigation. While current follow-up data extending beyond five years is encouraging, understanding whether single-dose gene therapy provides lifelong benefit or requires periodic re-dosing remains important. Some patients may experience gradual decline despite initial treatment response, necessitating strategies for re-dosing or alternative approaches.
The high cost of gene therapy presents significant economic and access challenges. While single-dose treatments theoretically offer cost-effectiveness compared to lifetime supportive care, upfront expenses exceed typical insurance coverage thresholds. Policymakers, healthcare systems, and manufacturers must collaborate to establish sustainable reimbursement models ensuring patient access while maintaining incentives for continued innovation.
Immunogenicity represents an additional consideration, as some patients develop immune responses to AAV vectors or transgene products. Research into immune tolerance strategies and immunosuppressive regimens aims to overcome these limitations. Understanding individual genetic and immunological factors predicting treatment response would enable personalized medicine approaches optimizing outcomes for each patient.
Recent Nature Medicine publications have highlighted emerging gene therapy approaches for lysosomal storage disorders, providing comprehensive reviews of mechanisms, clinical outcomes, and future directions. These peer-reviewed syntheses offer evidence-based perspectives on the field’s trajectory.
International collaboration and data sharing accelerate progress in Sanfilippo syndrome treatment. Patient registries, multicenter clinical trials, and open-access research platforms enable researchers worldwide to contribute insights and learn from collective experience. This collaborative approach has compressed timelines for therapeutic development and improved outcomes for affected patients globally.
Future innovations may include combination approaches pairing gene therapy with neuroprotective agents, anti-inflammatory interventions, or mitochondrial support strategies. Gene therapy combined with intensive rehabilitation, specialized education, and family support services creates comprehensive treatment packages addressing the multifaceted challenges Sanfilippo syndrome presents. This holistic approach recognizes that optimal outcomes require coordination across medical, therapeutic, educational, and social domains.
FAQ
What is the difference between gene therapy and enzyme replacement therapy for Sanfilippo syndrome?
Gene therapy delivers therapeutic genes that enable patients’ cells to produce deficient enzymes, potentially providing lifelong benefit from single or limited administrations. Enzyme replacement therapy involves regular infusions of manufactured enzymes, requiring ongoing treatment but avoiding genetic modification. Gene therapy offers superior CNS penetration and durability, while ERT provides more immediate effects with established safety profiles. Many experts view these approaches as complementary rather than competitive.
At what age can children receive gene therapy for Sanfilippo syndrome?
Current clinical trials primarily enroll young children, typically between ages one and six, before extensive neurological damage occurs. Earlier treatment generally produces better outcomes, making early diagnosis and treatment initiation critical priorities. Some research suggests preventive treatment of presymptomatic infants identified through newborn screening could optimize long-term outcomes, though this approach remains investigational.
How long do the effects of gene therapy last?
Current follow-up data extending beyond five years demonstrates sustained benefit in treated patients, with disease stabilization or improvement persisting throughout extended observation periods. Whether single-dose gene therapy provides lifelong benefit or eventually requires re-dosing remains under investigation. Individual variability in treatment response suggests some patients may require additional interventions, though current evidence supports durable disease modification in most treated individuals.
What are the side effects and risks of gene therapy?
Gene therapy generally demonstrates favorable safety profiles in clinical trials, with manageable adverse events. Common side effects include transient fever, headache, and mild CSF pleocytosis following intrathecal administration. Immune responses to AAV vectors occur in some patients but typically resolve with supportive care. Serious adverse events remain rare, though long-term safety monitoring continues as more patients receive treatment.
How is gene therapy administered to patients?
Current Sanfilippo syndrome gene therapies employ intrathecal injection, where therapeutic vectors are administered directly into cerebrospinal fluid surrounding the spinal cord and brain. This approach achieves high local CNS concentrations while minimizing systemic exposure. The procedure resembles lumbar puncture and typically requires hospitalization for monitoring. Emerging approaches may eventually enable less invasive administration routes, though intrathecal delivery remains the most clinically advanced approach.